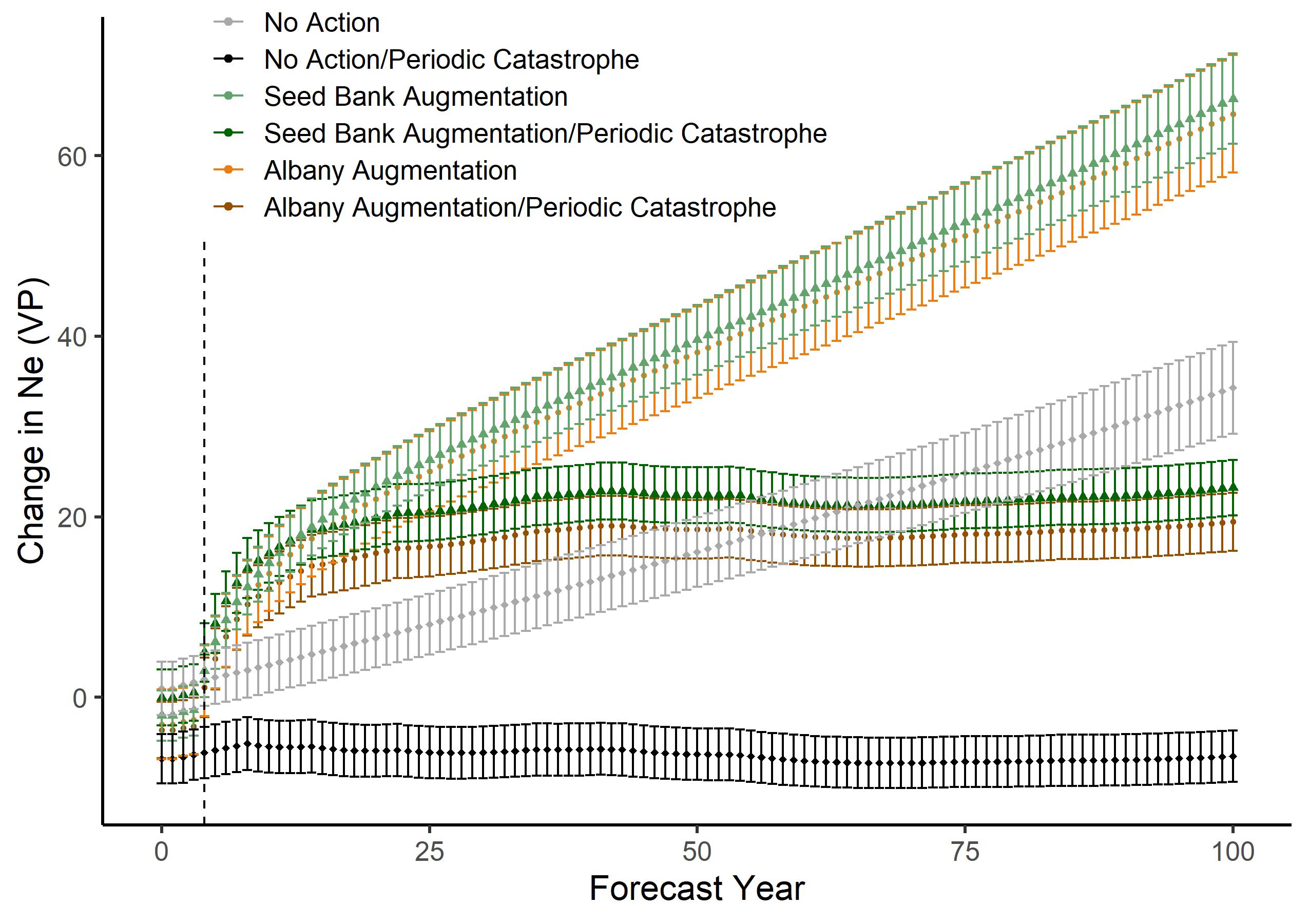
Sundial Lupine Assisted Gene Flow
Pine barren ecosystems are globally rare, among the most biodiverse areas in the temperate United States, and on the brink of disappearance. The pine barren legume Lupinus perennis (sundial lupine) serves as host plant to over a dozen rare insect species, including the federally endangered Karner Blue Butterfly and state threatened Frosted Elfin Butterfly. When restoring pine barren species, managers must first establish strong plant populations. Traditional wisdom states that “local is best” when sourcing seeds for a restoration, but this advice fails to consider the adverse effects of propagating inbred plants with bottlenecked diversity. We worked with state and nonprofit ecosystem managers to sequence seven Northeast sites, representing the majority of extant L. perennis populations that are under conservation protection or have seed bank accessions in the region. We used a ddRAD method to obtain a filtered set of 46,000 SNPs. Population area did not predict observed population heterozygosity (p = 0.36) or inbreeding (p = 0.42). A mantel test with Pearson’s correlation indicates population isolation by geographic distance (r = 0.754, p = 0.001). We modeled the genetic effects of translocating seeds from 30 year old seed bank accessions and from the geographically closest population for the Vermont population. We find that seed translocations greatly benefit genetic diversity and effective population size in this population, while inaction will lead to likely extirpation of this species in the state of Vermont. Our results illustrate the complexity in selecting a healthy restoration source population and suggest that the “local is best” adage does not match practical reality. Dividing populations by political boundaries, especially when those boundaries arbitrarily divide historically connected ecosystems, should not be taken as a one-size fits all solution for restoration.
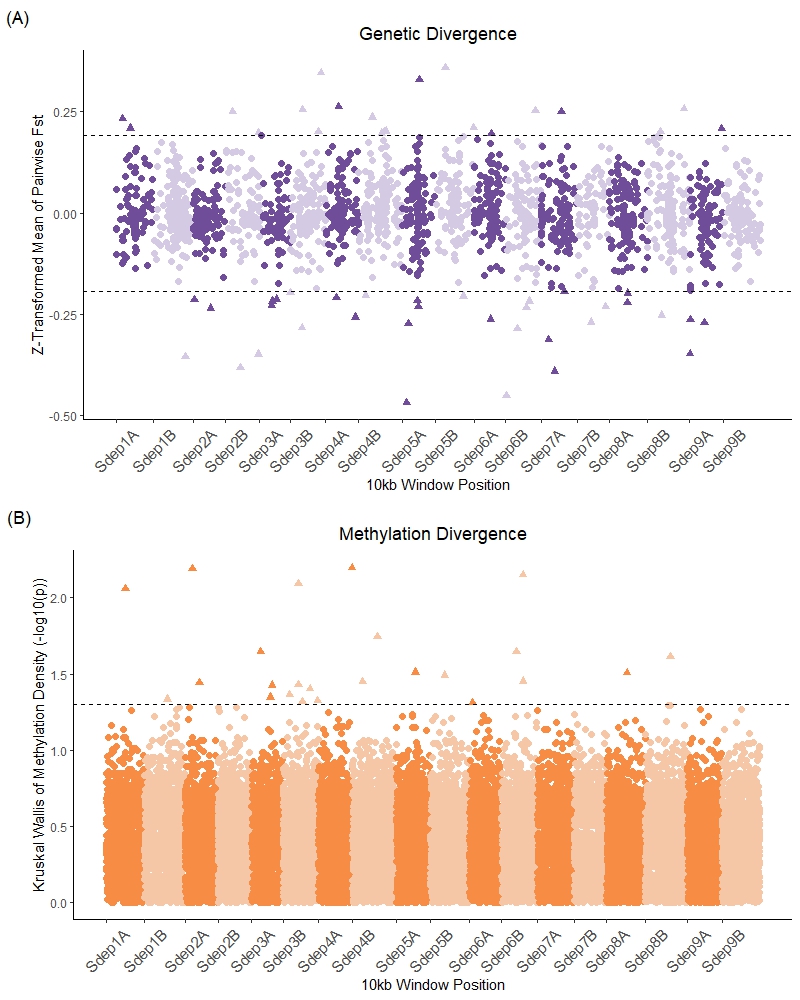
Salicornia Population Genomic and Methylation Variation
Methylation of CG motifs in many organisms is heritable, epimutable, and contributes to gene expression modulation. In plants, the rate of methylation epimutation is one million times faster than genetic mutation, making it one potential mechanism for rapid responses to the environment. Here we present a cost-effective approach using long-read Nanopore sequencing to simultaneously assess genetic and methylomic divergence and diversity in natural populations and demonstrate its utility in four populations of the the salt marsh plant Salicornia depressa. We find that pairwise genetic kinship between individuals is correlated with methylation profile (ρ = 0.3, p = 0.01). The four populations have similar genetic and methylation diversity across genomic windows, but highly genetically variable windows are uncorrelated with windows with high methylation density variation. Population divergence tests indicate that no genome windows are divergent in both genotype and methylation, as expected statistically if the two divergence types are uncorrelated. Our results suggest that regions of important genetic and methylation variation across populations are physically decoupled. Therefore, studies of complex trait evolution must consider the combined contributions of distinct genetic and methylation polymorphisms. Nanopore sequencing offers a promising avenue to reduce costs of such studies by obtaining genetic and methylation data concurrently. This approach could be a powerful tool to study species that lack standing genetic diversity, including crops and many plants of conservation concern. To encourage adoption and fill the gap of analysis tools available for Nanopore methylation data, we provide custom R scripts used for all methylation analyses.
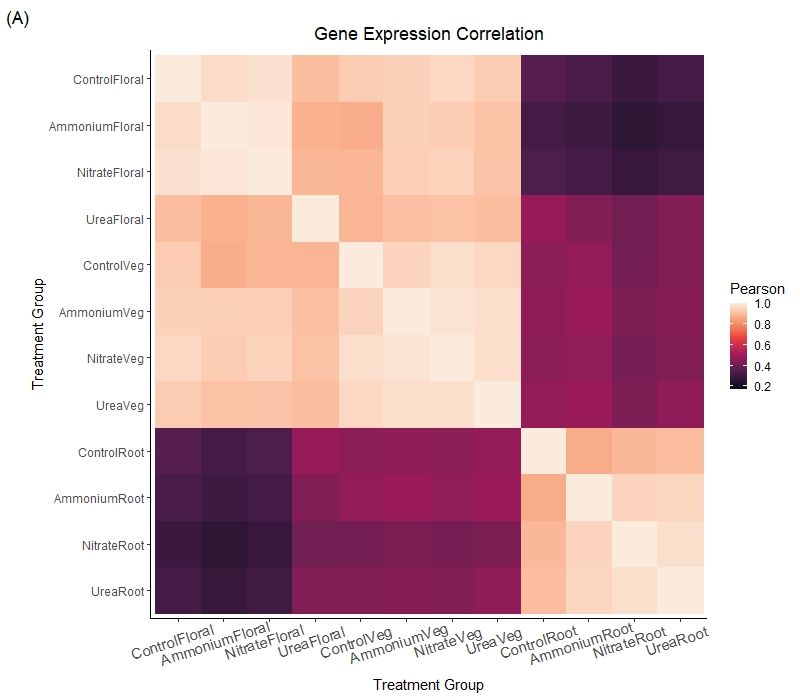
Salicornia depressa Transcriptomic Response to Nitrogen Stress
Salt marsh plants disproportionately depend on sub-species diversity for adaptation to global change stressors like nitrogen eutrophication. To characterize the response processes of the salt marsh plant Salicornia depressa, we carried out a controlled green house experiment with common nitrogen pollutants. We harvested root, shoot, and floral tissue from plants grown in control, ammonium, nitrate, and urea treatments. We sequenced RNA from each tissue sample and analyzed transcription profiles for tissue and treatment-specific differential gene expression. We found that ammonium treatment, which is associated with agricultural runoff, has the greatest effect on plant root to shoot biomass ratio and on transcription. Several of the ten most differentially expressed genes by nitrogen treatment are directly associated with nitrogen transport and metabolism functions, including the nitrate transporter NRT2.5 and the ammonium transporter AMT1. Two of the most differentially expressed genes have uncharacterized orthologues in Arabidopsis that have been linked to recent research on Universal Stress Proteins in other systems.
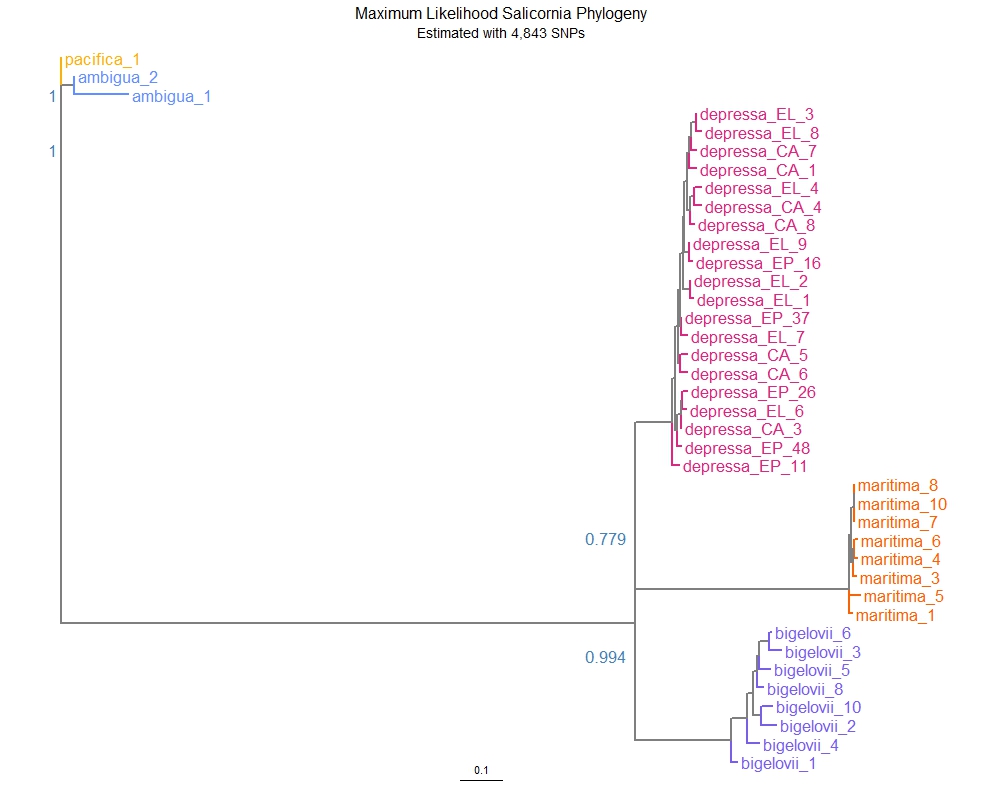
Salicornia Species Phylogenetics
Members of the Salicornia genus are infamously difficult to tell apart, leading to the Taxonomic Nightmare that preceded the last microsatellite marker-based species phylogeny. Following the 2007 reassignment, however, many marsh managers continue to struggle discerning plastic phenotypes from distinct species. To determine if the current taxonomic keys accurately describe Salicornia in their natural habitats, we collected samples of five purported species (ambigua, bigelovii, depressa, maritima, and pacifica) and sequenced them with a reduced representation (GBS) protocol. We used the denovo-based STACKS pipeline to call 4,843 SNPs with a 40% missingness filter from our multi-species sequencing library. A PCA shows clear separation of assigned species groups along the first three principal components. We then constructed a maximum likelihood phylogeny with RAxML, which showed complete monophyly for the samples of each species. Notably, S. depressa samples taken from both the east and west coast of North American grouped together over other species cohabiting the same sample sites. Our results show strong support for the current phylogenetic grouping and taxonomic guidance for these five species. Further analysis of alignment rates against the Salicornia depressa reference genome support the assignment of chromosomes to their allotetraploid subgenomes. All samples of the diploid S. maritima show greater alignment to the A subgenome (p < 0.0001) and nearly zero alignment to the B subgenome, suggesting the former is of North American lineage while the latter may originate from diploid species in Europe.

Genome Assembly
Salicornia depressa (American pickleweed) is the most widespread member of its salt-loving genus in North America. Pickleweeds typically colonize high marsh areas that tides make highly saline. Most other salt marsh plants cannot withstand the same salt pressure, so these bare patches are free from competitive pressures. Understanding the genetic origin of American Pickleweed’s high salinity adaptations has potential application to salt-tolerant agriculture and salt marsh conservation but requires genome resources to study. Salicornia depressa is a tetraploid species, which presents unique challenges to traditional genome assembly pipelines. We present a high-quality, chromosome-scale reference genome of S. depressa and the pipeline we used to assemble it. Our reference is phased to each of the tetraploid’s ancestral subgenomes with a scaffold N50 of 69.3 Mb, BUSCO completeness of 96.4%, and k-mer completeness of 98.4%. The subassemblies are evenly split, with subassembly A containing 56.7% and subassembly B containing 59.1% of genomic k-mers. Our gene annotation identifies 80,883 genes and our methylome annotation contains 2.5 million methylated cytosines. We find that gene and methylation density are negatively correlated across the genome. We also assembled and annotated a chloroplast assembly which includes all expected photosystem, tRNA, and rRNA genes. We provide a guide to our successful assembly pipeline involving >30 programs. Our reference and annotation join resources for three other Salicornia species, allowing global scale ecological-evolutionary studies.
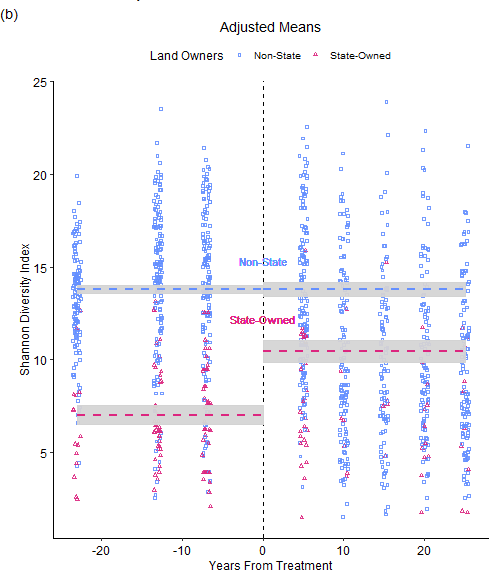
Policy Analysis
Many forestry policies aim to balance economic use with conservation. Considering the combined human and ecological dimensions of these policies can provide key insight into the real-world effects of management intervention changes. Counterfactual study designs approach this problem by considering what would have happened without management changes. We take a counterfactual approach to assess the 1996 amendment to the North Carolina Forestry Council, which for the first time required state forest best management practices to consider factors other than profit. We hypothesize that tree biodiversity in North Carolina’s state forests today is greater than it would be without this 1996 amendment. We construct counterfactual control units from USDA Forest Inventory and Analysis survey data of privately-owned forests, which were not affected by the policy change. We aggregated and rarefied plot species counts based on ownership status to calculate Shannon Diversity Indices at the county level over a fifty-year period. Our study design is supported both theoretically and empirically by a parallel trend test. A Difference-In-Difference model estimates the Average Treatment effect on the Treated of a 3.994 increase in Shannon diversity (p = 0.001) with a 95% confidence interval of 2.162 to 5.825. Our assessment also highlights the negative biodiversity trends on privately-owned lands. Future research should aim to assess and synthesize policy impacts to identify approaches that balance the economic and environmental aspects of forest management.